Why start from scratch with Bayesian optimization for every new reaction?
Most Bayesian optimization in chemistry still starts from scratch for every new reaction. It’s an expensive way to pretend we’re starting from zero.
Transfer learning lets models start smarter. Instead of rediscovering basic trends in reactivity, solvent effects, or catalyst performance, models can use prior experiments as a foundation. The real question isn’t whether to transfer knowledge, but how to do it without being pulled in the wrong direction by mechanistic differences between systems.
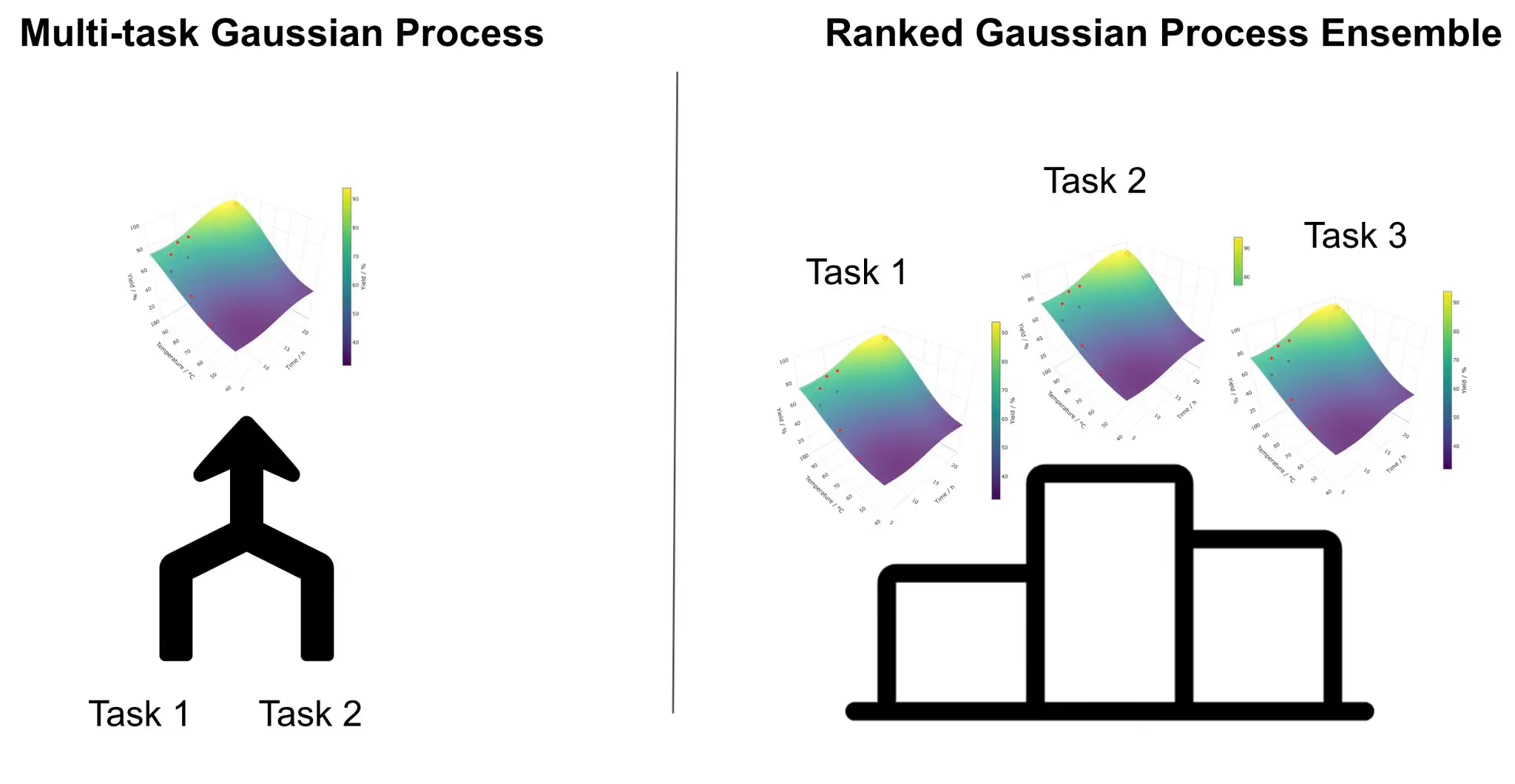
Multi-Task Gaussian Processes (MTGPs) work well when you believe the chemistry is highly correlated. They assume multiple tasks share a common structure and learn a shared representation across them. This coupling is powerful when the assumption holds, but when the systems diverge - different substrates, different rate-determining steps, different failure modes - negative transfer may occur, slowing optimization rather than accelerating it.
Ranked Gaussian Process Ensembles (RGPEs) take a different view. Instead of enforcing shared structure, they compare models trained on prior datasets to the one being optimized and weight them based on how well they predict the new system. Prior data is reused when it is useful and quietly ignored when it isn’t. This makes RGPEs far more robust when the data is messy, the chemistry is varied, and you don’t fully trust your assumptions.
The rule of thumb is simple: rely on MTGPs only when you truly trust similarity across systems. When you don’t, let a method like RGPE decide how much past chemistry to reuse. This is the principle behind MemoryBO at ReactWise- transfer, but only when it helps.
We'll be sharing our results at the Active Learning Symposium today with case studies from pharma and our own proprietary data. I look forward to seeing you there.



